
Con il termine neurofibromatosi si intendono un gruppo di patologie genetiche tutte caratterizzate dalla presenza di neurofibromi o neuromi. Ciò che le distingue è la patogenesi, la severità del quadro clinico e la prognosi. Le neurofibromatosi sono classificate come malattie rare. La forma più frequente (85% di tutti i casi diagnosticati) è la NF1 con un’incidenza intorno a 1:3000 nuovi nati. È trasmessa con modalità autosomica dominante nelle forme familiari, ma in un 50% di casi si osservano mutazioni spontanee del gene.
Classificazione delle neurofibromatosi
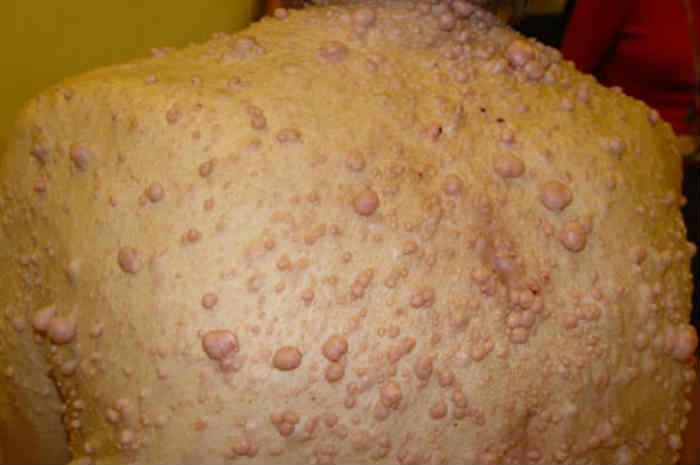
Manifestazione di neurofibromatosi
Inizialmente le neurofibromatosi sono state classificate in base alla clinica. Successivamente, la ricerca sulla genetica di queste patologie ha permesso di individuare due entità distinte: la neurofibromatosi tipo 1 (NF1) detta anche malattia di Von Recklinghausen o neurofibromatosi periferica, e la neurofibromatosi tipo 2 (NF2) o neurofibromatosi centrale o BANF (bilateral acoustic neurofibroma).
Nel 1982 Riccardi classifica altre 6 forme di neurofibromatosi, basate solo sulla clinica. Nel 1992 ne sono state riconosciute altre tre forme.
| Classificazione delle neurofibromatosi | |
| Riccardi - Eichner, 1982 | Neurofibromatosi tipo 1 (NF1 o malattia di Von Recklinghausen) Neurofibromatosi tipo 2 (NF2 o BANF: bilateral acoustic neurofibroma) Neurofibromatosi tipo 3 (NF3 o neurofibromatosi mista) Neurofibromatosi tipo 4 (NF4 o neurofibromatosi variante) Neurofibromatosi tipo 5 (NF5 o neurofibromatosi segmentale) Neurofibromatosi tipo 6 (NF6) solo macchie Neurofibromatosi tipo 7 (NF7 ad esordio tardivo) Altre Neurofibromatosi (NOS: non altrimenti specificate) |
| Riccardi - 1992 | Neurofibromatosi gastro-intestinale Schwannomatosi multipla Tumori cerebrali multipli |
Neurofibromatosi Tipo 1
I neurofibromi sono formazioni color carne che originano dalle cellule di Schwann (che avvolgono le fibre nervose periferiche) e da altre cellule di sostegno dei nervi periferici.
Le neurofibromatosi sono classificate come malattie rare. La forma più frequente (85% di tutti i casi diagnosticati) è la NF1 con un’incidenza intorno a 1:3000 nuovi nati. È trasmessa con modalità autosomica dominante nelle forme familiari, ma in un 50% di casi si osservano mutazioni spontanee del gene. La gravità del disturbo dipende dal tipo di mutazione.
Manifestazione di neurofibromi
La diagnosi di NF1 si formula quando c’è la presenza di due o più delle seguenti alterazioni:
- macchie della pelle di colore marrone chiaro (dette “macchie caffellatte”) in numero minimo di 6
- neurofibromi (in numero minimo di 2)
- lentiggini alle ascelle o all’inguine
- noduli di Lisch in numero superiore o uguale a 2
- glioma del nervo ottico
- sviluppo anomalo della colonna vertebrale o dello sfenoide o della tibia
- un familiare di primo grado affetto da NF1
I pazienti affetti da questa patologia possono presentare anche disturbi dell’apprendimento, lieve ritardo cognitivo, convulsioni, ipertensione arteriosa.
Inoltre presentano un 10% di rischio in più di sviluppare altri tumori maligni a carico della guaina mielinica, della mammella, feocromocitoma, rabdomiosarcoma, leucemia.
Andamento della Neurofibromatosi Tipo 1
La NF1 è solitamente asintomatica, escludendo le macchie caffellatte e i neurofibromi sottocutanei.
Se però questi ultimi esercitano una pressione sul nervo che circondano, i soggetti possono accusare parestesie o deficit di forza nelle aree interessate dal nervo.
I neurofibromi possono interessare qualsiasi nervo del corpo, ma si sviluppano più frequentemente sulle radici dei nervi spinali. Qui non determinano conseguenze rilevanti, tranne in caso di compressione del midollo spinale che si manifesta con paralisi e alterazione della sensibilità.
Generalmente la progressione della NF1 è lenta, ma con l’aumentare del numero di neurofibromi, le alterazioni nuerologiche si intensificano.
Attualmente non esiste una cura specifica per la NF1. Fondamentale è la tempestività della diagnosi per limitare le complicanze potenziali.
Essendo a tutti gli effetti una patologia multisistemica, è necessario un approccio terapeutico multidisciplinare per garantire la qualità del risultato.
Neurofibromatosi Tipo 2
È una condizione molto più rara rispetto alla NF1. Colpisce circa 1 persona su 25000 e il meccanismo di trasmissione è sempre su base genetica.
Caratteristica della NF2 è la presenza di tumori (detti schwannomi) a lento sviluppo, solitamente a carico dell’ottavo nervo cranico, cioè del nervo acustico. Ciò si manifesta con sintomi che riguardano la funzione uditiva e l’equilibrio, determinando forme anche gravi di disabilità.
Nei pazienti affetti da NF2 possono presentarsi anche altre tipologie di tumori del sistema nervoso come i meningiomi e i gliomi.
Nei bambini l’esordio dei sintomi può essere così lento da passare inosservato fino all’età di 20 anni e si manifesta con problematiche relative all’apparato uditivo.
Andamento della Neurofibromatosi Tipo 2
L’andamento della malattia varia notevolmente da un individuo all’altro e il peggioramento dei sintomi può essere lento, fino al quadro peggiore associato allo sviluppo di meningiomi. Anche per la NF2 non esiste una terapia mirata, ma la gestione integrata con approccio multidisciplinare costituisce la scelta migliore e, per ora, più efficace.
















Commento (0)
Devi fare il login per lasciare un commento. Non sei iscritto ?