
La sindrome del QT lungo o Long QT Syndrome (LQTS) è una patologia cardiaca, sia congenita che acquisita, che riguarda una anomalia dell'attività elettrica del cuore. È solitamente una malattia genetica ereditaria determinata da alterazioni elettrofisiologiche, con una gravità variabile a seconda del tipo di mutazione implicata. Può essere anche secondaria a fattori ambientali come l'assunzione di alcuni farmaci e disordini elettrolitici. Si verifica un allungamento dei tempi di recupero dei ventricoli del cuore, dopo una prima contrazione, nel prepararsi a quella successiva. Il cuore impiega più tempo del normale a rilassarsi fra una contrazione e l'altra, causando battiti veloci ed irregolari. E se il cuore batte in modo anormale per un tempo troppo lungo può provocare una morte improvvisa. La sindrome del QT lungo è la prima causa di morte improvvisa sotto i 20 anni.
Segni e sintomi della sindrome del QT lungo
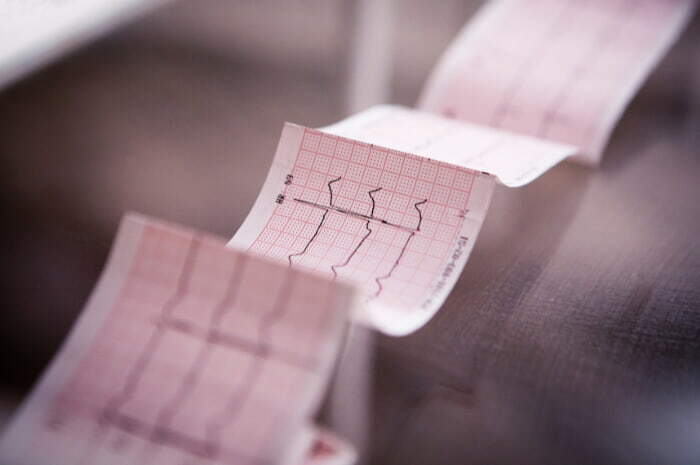
Le aritmie vengono diagnosticate analizzando il profilo del tracciato elettrocardiografico, sono denominate secondo il punto di origine dell'impulso e il meccanismo di formazione o di conduzione implicati.
La condizione, rara ma ben nota, è caratterizzata dal prolungamento dell'intervallo QT e da anomalie morfologiche dell'onda T.
Si tratta di alterazioni di parametri specifici dell'elettrocardiogramma, l'esame strumentale che registra l'attività elettrica del cuore, che aumentano il rischio di insorgenza di aritmie maligne potenzialmente letali – come una tachicardia ventricolare tipo torsione di punta (grave compromissione della contrazione cardiaca che riduce l'afflusso di sangue al cervello) e una fibrillazione ventricolare (profonda alterazione della frequenza cardiaca).
Tali complicanze possono provocare episodi sincopali (perdita di coscienza immediata), arresto cardiaco e morte improvvisa.
La sindrome è solitamente asintomatica. I segni e i sintomi più comuni - come svenimenti, palpitazioni, sensazioni di mancamento e crisi epilettiche – si manifestano senza segni prodromici. Lo svenimento è causato dalla contrazione troppo rapida e non efficace dei ventricoli.
L'onda elettrocardiografica rappresenta la funzione del sistema di conduzione, che normalmente dà inizio e propaga l'attività elettrica del cuore, e viene registrata come un tracciato su una carta millimetrata, suddivisa in intervalli di dimensioni standard da linee verticali e orizzontali più o meno marcate.
L'asse orizzontale del grafico rappresenta il tempo o la velocità, l'asse verticale l'ampiezza o il voltaggio. Quando il profilo dell'onda ECG si sposta verso l'alto della carta si parla di deflessione positiva, quando si sposta verso il basso si parla di deflessione negativa
L'elettrocardiogramma (ECG) è composto da onde (un'onda P, un complesso QRS, un'onda T e, talvolta, un'onda U) e da segmenti o intervalli (intervallo PR, segmento ST, intervallo QT). L'intervallo PR viene misurato dall'inizio dell'onda P all'inizio del complesso QRS; QRS dall'inizio dell'onda Q alla fine dell'onda S; l'intervallo QT dall'inizio dell'onda Q alla fine dell'onda T.
L'intervallo QT – che si misura dall'inizio del complesso QRS alla fine dell'onda cardiaca - è pertanto un elemento del tracciato elettrocardiografico corrispondente al tempo che intercorre tra la depolarizzazione e la ripolarizzazione ventricolari. La depolarizzazione o stimolazione elettrica (fase di contrazione del cuore) è il processo durante il quale una cellula muscolare cardiaca passa da una condizione in cui l'ambiente intracellulare è carico negativamente rispetto all'ambiente extracellulare ad una in cui è carico positivamente.
La ripolarizzazione o rilassamento elettrico è il processo durante il quale una cellula muscolare ritorna alla condizione di riposo in cui l'ambiente intracellulare è carico negativamente rispetto all'ambiente extracellulare. Depolarizzazione e ripolarizzazione di atri e ventricoli hanno tempi specifici, se la ripolarizzazione è ritardata si originano aritmie.
Per generare un battito cardiaco normale è necessario che ci sia un corretto passaggio di cariche elettriche – condotte da ioni sodio, potassio e calcio - tra l'esterno e l'interno delle cellule cardiache. Se tali correnti ioniche sono alterate, le alterazioni generano un marcato allungamento del tempo elettrico necessario al cuore per ritornare, dopo ogni battito, nella condizione di riposo. Se il tempo di attivazione elettrica si allunga, l'intervallo QT si prolunga e si generano aritmie.
Le aritmie sono anomalie della formazione e/o della conduzione di un impulso elettrico a livello cardiaco. Esse possono causare disturbi della frequenza cardiaca, del ritmo cardiaco o di entrambi. Possono rivelarsi inizialmente attraverso il loro effetto emodinamico, vengono diagnosticate analizzando il profilo del tracciato elettrocardiografico e sono denominate secondo il punto di origine dell'impulso e il meccanismo di formazione o di conduzione implicati.
Le forme genetiche della sindrome del QT lungo – le due principali sono la Romano Ward e la Jervell Lange Nielsen - si manifestano prevalentemente in età pediatrica ma possono esordire anche dopo la pubertà. Hanno un'incidenza di circa 1 persona ogni 2000.
Derivano da una canalopatia ossia da disturbi genetici della funzione o della regolazione dei canali ionici che prolungano la durata del potenziale di azione del miocita ventricolare (il potenziale di azione è la rapida variazione del potenziale di membrana, che passa da un valore negativo di riposo - circa -90mV - ad un valore positivo per poi ritornare al valore iniziale). Questa disfunzione aumenta il rischio che si verifichino oscillazioni di tensione tra le membrane durante il potenziale. Pertanto, se in una determinata area locale le durate del potenziale dei miociti variano, tali oscillazioni possono riattivare i miociti vicini che si sono ripolarizzati.
Diagnosi di sindrome del QT lungo
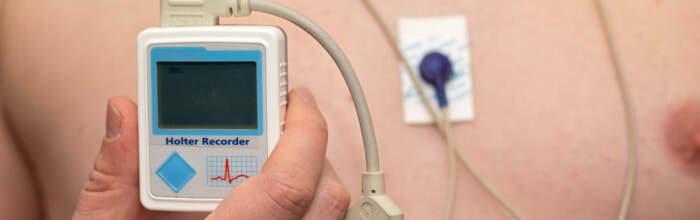
Nella diagnosi occorre individuare le cause acquisite, modificabili e verosimilmente curabili. La diagnosi di LQTS è basata principalmente sull'ECG eseguito a riposo o sotto sforzo. Poiché alcune anomalie compaiono solo durante l'esercizio, l'ECG sotto sforzo consente di individuare la sindrome anche in quei pazienti con un intervallo QT borderline.
Il riscontro può essere occasionale oppure in corso di tachicardia ventricolare documentata in assenza di altre cause di intervallo QT prolungato. Il monitoraggio ambulatoriale dell'ECG con Holter può rilevare alterazioni transitorie della ripolarizzazione ventricolare consentendo la valutazione delle modificazioni dinamiche dell'intervallo QT e dell'onda T.
A volte può essere necessario, per rilevare un QT lungo nascosto in pazienti con intervallo QT normale ma con una buona probabilità di essere affetti dalla sindrome congenita, attuare il test provocativo somministrando isoproterenolo EV o adrenalina.
Come si tratta la sindrome del QT lungo
La sindrome è curabile. Le possibilità di cura attualmente disponibili sono i farmaci beta bloccanti, preferibilmente a lunga durata d'azione e a lento rilascio. Pur non modificando il QT troppo lungo, essi permettono di proteggere in maniera aspecifica dalle aritmie e riescono a prevenire la maggior parte dei sintomi.
La risposta terapeutica è tuttavia variabile, la loro efficacia dipende dal tipo di difetto genetico coinvolto e dalla durata dell'intervallo QT. Se la risposta ai betabloccanti non è sufficiente e le aritmie sono gravi, può rendersi necessario ricorrere in aggiunta all'impianto di un defribillatore automatico (ICD, Impiantable Cardioverter Defribillator). Si tratta di una terapia non farmacologica con un dispositivo salvavita che controlla il ritmo cardiaco ed interviene in caso di aritmie ventricolari pericolose.
La tachicardia ventricolare tipo torsione di punta può essere trattata con la cardioversione e il solfato magnesio EV. È possibile accorciare l'intervallo QT aumentando la frequenza cardiaca, nei pazienti con frequenti o lunghi cicli di tachicardia ventricolare tipo torsione di punta, tramite stimolazione temporanea (manovra di rianimazione tramite sonda), isoproterenolo o entrambi.
La stimolazione permanente tramite pacemaker (stimolatore cardiaco impiantato tra la cute e il muscolo pettorale), utile per aumentare la frequenza ventricolare basale e per prevenire le pause post-extrasistoliche, può ridurre la probabilità di TV tipo torsione di punta.
Nei casi più problematici si può attuare, anche se raramente, la denervazione simpatica cardiaca sinistra: consiste nell'asportazione chirurgica delle terminazioni nervose responsabili di alterazioni del ritmo. Ci sono infatti alcuni gangli localizzati in alcune aree del torace superiore che, se stimolati, possono dare origine ad una marcata instabilità elettrica.















Commento (0)
Devi fare il login per lasciare un commento. Non sei iscritto ?